- Kategorie: Programme Area 4
Immuno-Epigenetics

- Kategorie: Cell therapy
- Kategorie: Epigenetic imprinting
- Kategorie: Epigenetic Regulation
- Kategorie: T-Lymphocytes
Understanding the epigenome in T cells to develop new therapies
For many rheumatic diseases, curative therapies are still missing, leaving patients dependent on lifelong immunosuppressive treatment. Adoptive cell therapies offer a promising strategy for curative therapies with the goal of achieving full immunological homeostasis – either by permanently enhancing the immunosuppressive arm of the immune system (i.e. Treg therapy) or by eliminating the pathogenic autoreactive part (e.g. CAR T cell therapy).
Our research group investigates the epigenetic mechanisms governing T cell differentiation, function, and senescence in both health and disease. From the knowledge gained, we aim at developing approaches for directed epigenetic interventions to generate new cell therapies for autoimmunity and chronic inflammatory diseases. With this, we make use of the regulatory potential, which is mediated by the 3-dimensional folding of the genome, to steer T cells towards optimal functional states for their use in cell therapy approaches.
To this end, our group utilizes a broad portfolio of techniques for the epigenetic profiling of both, whole genomes and specific target genes, which are applicable to low input samples such as clinical specimen. In addition, our group develops and applies innovative approaches for the targeted modification of epigenomic structures, to be used to optimize therapeutic cell products.
By integrating fundamental epigenetic research with applied therapeutic strategies, our group seeks to advance the development of epigenetically engineered cell therapies for immune-mediated diseases.


Our Research
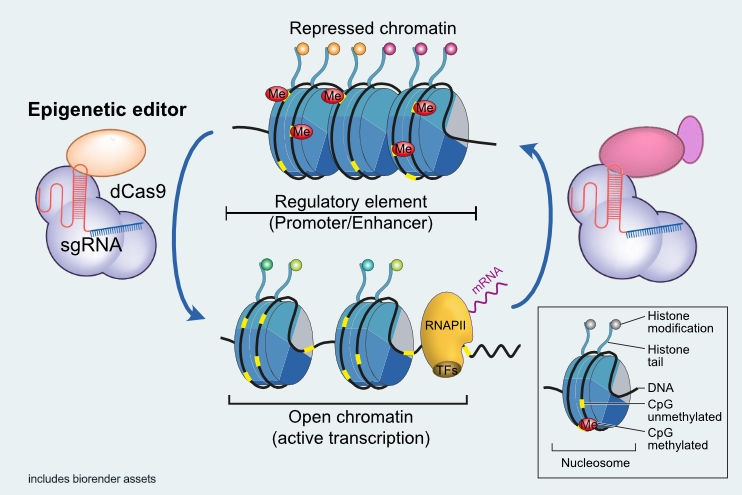
Epigenetic modifications on regulatory elements in the genome control which genes are currently expressed, which genes will be expressed in response to signals, and which are stably silenced. Thus, they act as molecular switches, deciding whether genes are turned “on” or “off”. While such regulatory elements can be identified based on evolutionary DNA sequence conservation and differential epigenetic states, elucidating the functional contribution of individual epigenetic marks to gene expression control remains challenging and often limited to correlative observations.
To address this challenge, we established CRISPR/Cas-based epigenetic editing methods, with which we are able to selectively modify epigenetic marks at specific genomic loci. With this, we can assess the functional role of the targeted epigenetic mark on a selected regulatory element in the native chromatin surroundings of a living cell.
Beyond fundamental research, epigenetic editing holds immense promise for therapeutic applications, such as during adoptive T cell therapy which is currently also tested for application in rheumatic diseases. Here, epigenetic editing can be used to equip therapeutic T cell products with desired qualities, such as migration properties, resilience against inhibitory checkpoint molecules or pro-inflammatory and anti-inflammatory functions as required.
enlarge image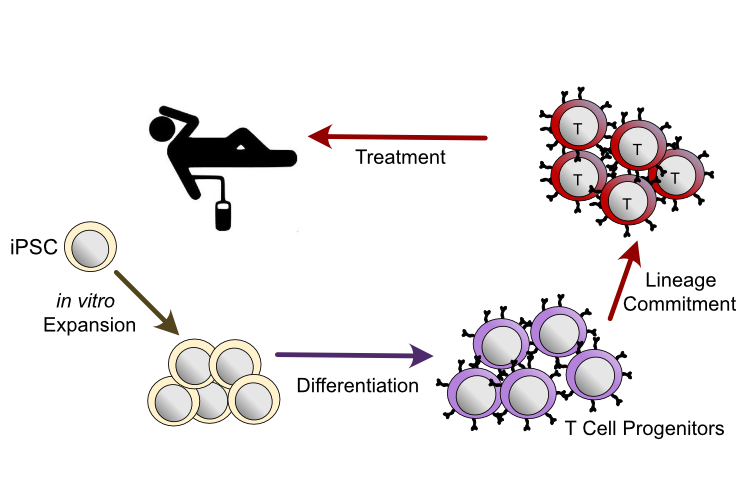
T cells are a highly specialized immune cell subset which are central to the body’s adaptive immune response. Currently their use in adoptive T cell therapy – a form of personalized immunotherapy – to target diseases from cancer to autoimmunity and including rheumatic diseases is intensively investigated worldwide. However, a major limitation is the availability of these T cell products in adequate numbers.
Thus, we investigate the possibilities to generate different T cell populations in the lab (=in vitro) from a replenishing source of induced pluripotent stem cells (iPSCs). We work with state-of-the-art techniques to mimic the development of immune cells in the human body and compare our in vitro generated T cell progenitors to their counterparts in the human body on a phenotypical and epigenetic level.
enlarge image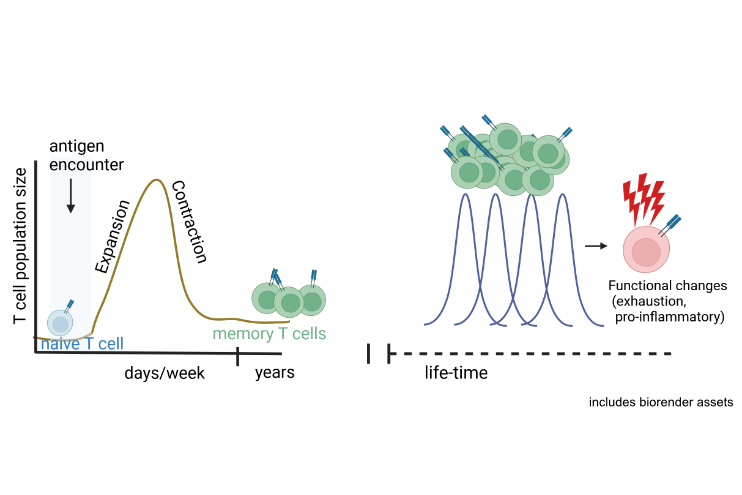
Memory T cells differentiate from antigen-experienced T lymphocytes and persist long-term, conferring immunological protection by orchestrating a rapid and robust response upon re-exposure to previously encountered pathogens. Due to their antigen specificity, T cells must undergo extensive clonal expansion to generate a sufficient number of effector cells for effective pathogen elimination. As long-lived immune cells, memory T cells must safeguard their functional identity despite repeated proliferative cycles. This immunological identity is governed by the epigenome, a complex network of chemical modifications that regulate chromatin structure and gene expression. Among these, DNA methylation and histone modifications are the best-studied epigenetic marks, controlling chromatin accessibility and thereby shaping the cell type-specific transcriptional landscape. The preservation of epigenetic integrity is paramount to sustaining the stability and effector potential of memory T cells, allowing them to mediate durable immune surveillance for decades.
Emerging evidence indicates that under conditions that promote proliferation, such as inflammatory microenvironments, T cells may encounter challenges in preserving their cellular identity, potentially resulting in functional alterations. This phenomenon is not restricted to pathological contexts but is also of significant concern in therapeutic cell manufacturing, where extensive in vitro expansion is required. Prolonged in vitro culture has been associated with epigenetic and transcriptional changes, potentially compromising the stability, potency, and therapeutic efficacy of these cellular products.
DNA methylation maintenance in heterochromatin is imperfect during proliferative episodes, leading to progressive hypomethylation of normally highly methylated regions. This suggests that excessive cell division challenges epigenetic stability. One key aspect of our research is the study of proliferation-associated epigenetic changes and their functional impact on T cells. By unravelling these mechanisms, we aim to understand how proliferation shapes T cell identity and function in both health and disease and make use of the insights gained for the improvement of cell therapy approaches for rheumatic diseases.
enlarge image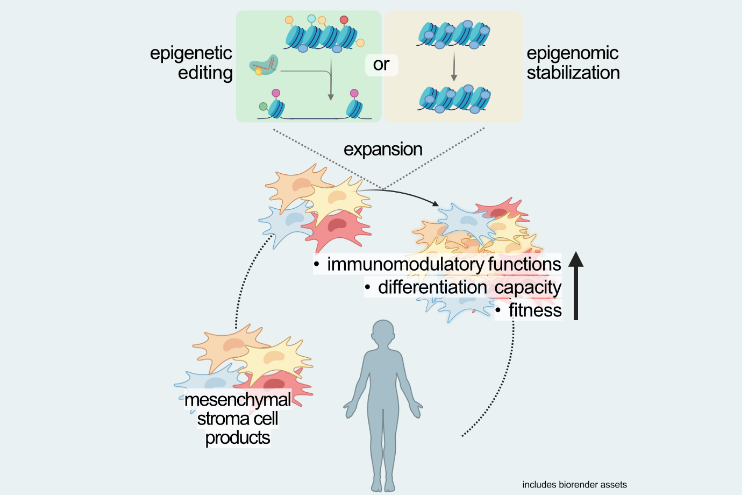
Mesenchymal stromal cells (MSCs) hold great promise for cell-based therapies due to their multipotent differentiation capacity, immunomodulatory functions, and accessibility from various tissue sources. Their versatility makes them an interesting cell type for research, with a prospect of potential therapies for inflammatory disorders and enhancing tissue regeneration, including cartilage regeneration in osteoarthritis.
This project focuses on translating our insights on epigenomic stability and modulation, gained through our research on lymphocytes, to a new cell type, MSCs, with the goal of further improving their therapeutic utility.
For this, we aim to better understand epigenetic regulatory mechanisms and patterns throughout cell differentiation of MSCs using in-depth comparative epigenomic profiling to identify critical regulatory sites that primarily drive the transcriptomic program during differentiation. Ultimately, our goal is to leverage our epigenetic editing approach to precisely activate regulatory elements, and with this, steer cellular fates in a targeted manner, with a particular focus on the in vitro differentiation capacity of MSCs into cartilage-like tissue.
In addition, we aim to counteract progressive DNA methylation changes observed during in vitro cell expansion, as these may impact the fitness and functionality of cell products.
enlarge image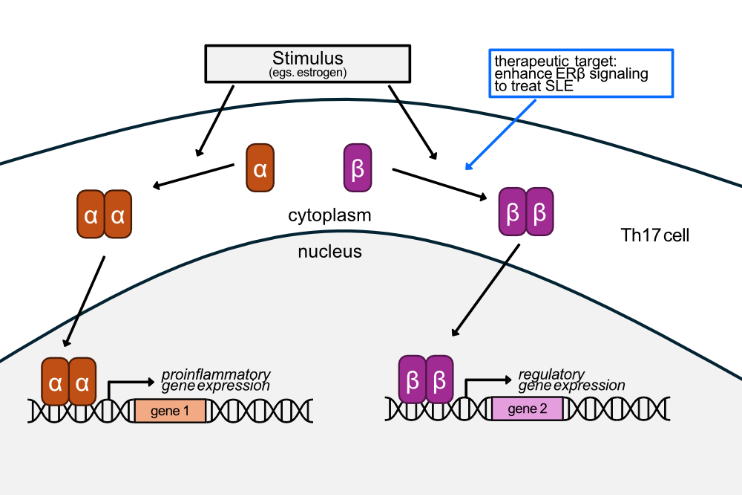
CD4+ T helper (Th) cells can contribute to the development of autoimmunity by orchestrating an inflammatory response to the body. Th17 cells are a pro-inflammatory subset of CD4+ Th cells that contribute to many autoimmune diseases, including Systemic lupus erythematosus (SLE). The Estrogen receptor ERβ is expressed in Th17 cells of people of all sexes, but its expression is decreased in the immune cells of many SLE patients. Our goal is to understand how ERβ plays a role in healthy Th17 cells, and how its loss might contribute to SLE.
enlarge image








